

Carbon nanostructures could become easier to design and synthesize thanks to a machine learning method that predicts how they grow on metal surfaces. The new approach, developed by researchers at Japan's Tohoku University and China's Shanghai Jiao Tong University, will make it easier to exploit the unique chemical versatility of carbon nanotechnology. The method was published in the journal Nature Communications.
The growth of carbon nanostructures on a variety of surfaces, including as atomically thin films, has been widely studied, but little is known about the dynamics and atomic-level factors governing the quality of the resulting materials. "Our work addresses a crucial challenge for realizing the potential of carbon nanostructures in electronics or energy processing devices," says Hao Li of the Tohoku University team.
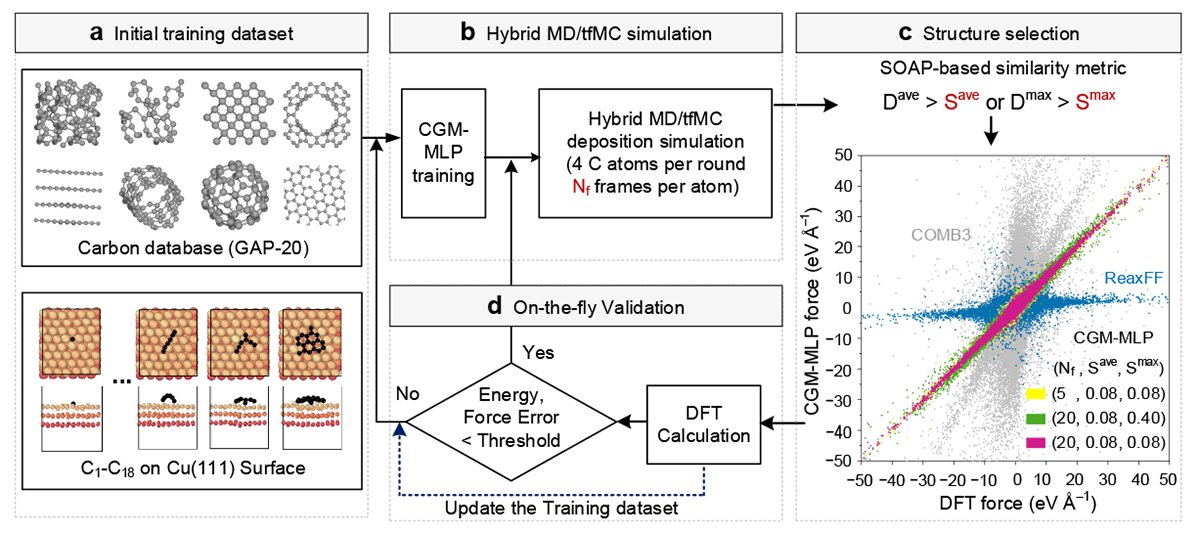
The wide range of possible surfaces and the sensitivity of the process to several variables make direct experimental investigation challenging. The researchers therefore turned to machine learning simulations as a more effective way to explore these systems.
With machine learning, various theoretical models can be combined with data from chemistry experiments to predict the dynamics of carbon crystalline growth and determine how it can be controlled to achieve specific results. The simulation program explores strategies and identifies which ones work and which don't, without the need for humans to guide every step of the process.
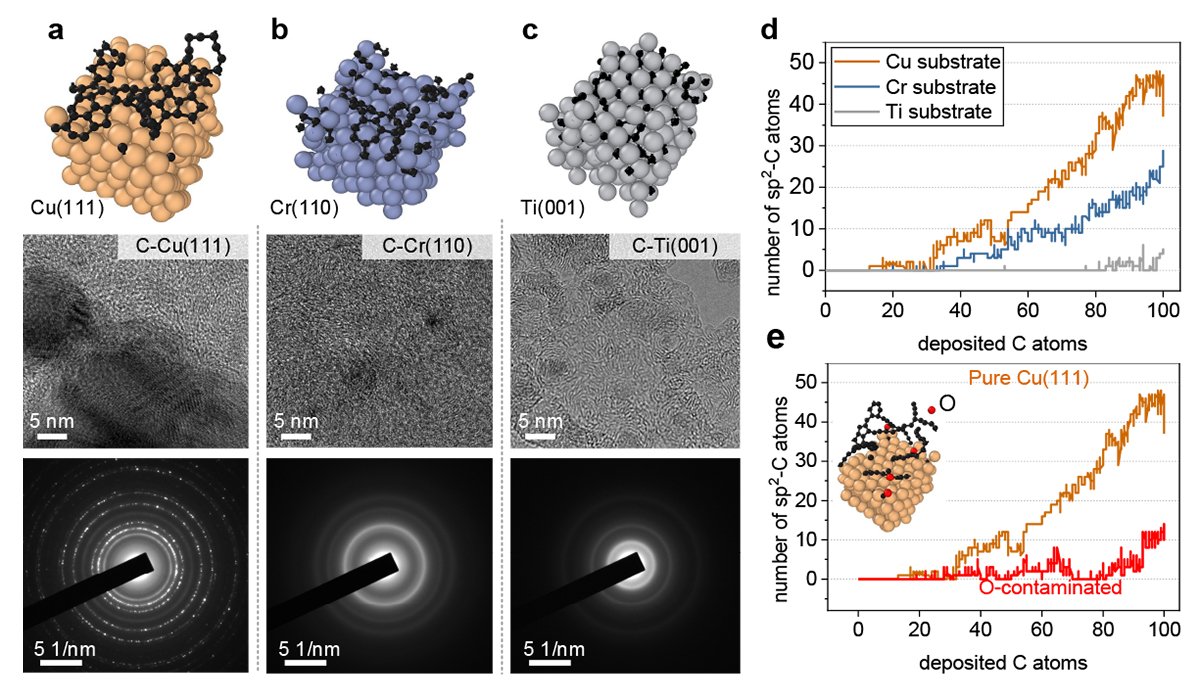
The researchers tested this approach by investigating simulations of the growth of graphene, a form of carbon, on a copper surface. After establishing the basic framework, they showed how their approach could also be applied to other metallic surfaces, such as titanium, chromium and copper contaminated with oxygen.
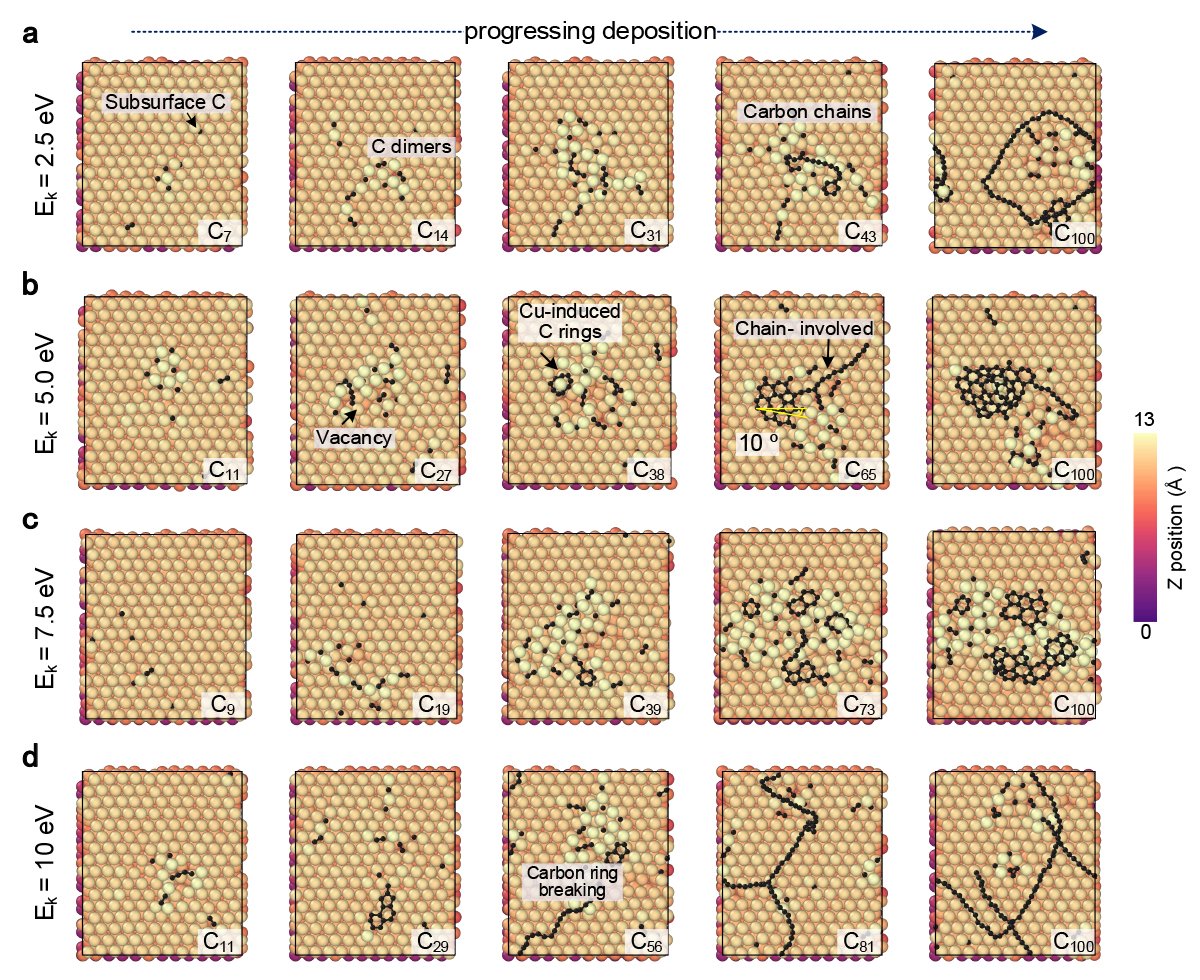
The distribution of electrons around the nuclei of atoms in different forms of graphene crystals can vary. These subtle differences in atomic structure and electron arrangement affect the overall chemical and electrochemical properties of the material. The machine learning approach can test how these differences affect the diffusion of individual atoms and bonded atoms and the formation of carbon chains, arches and ring structures.
The team validated the results of the simulations through experiments and found that they closely matched. "Overall, our work provides a practical and efficient method for designing metallic or alloy substrates to achieve desired carbon nanostructures and explore further opportunities," Li says.
He adds that future work will build on this to investigate topics such as the interfaces between solids and liquids in advanced catalysts and the chemical properties of materials used for processing and storing energy.
- Publication Details:
Title: Active Machine Learning Model for the Dynamic Simulation and Growth Mechanisms of Carbon on Metal Surface
Authors: Di Zhang, Peiyun Yi, Xinmin Lai, Linfa Peng, Hao Li
Journal: Nature Communications
DOI: 10.1038/s41467-023-44525-z
365体育|365体育投注@:
Hao Li,
Advanced Institute for Materials 365体育|365体育投注@ (WPI-AIMR), Tohoku University
Email: li.hao.b8 tohoku.ac.jp
tohoku.ac.jp
Website: https://www.li-lab-cat-design.com/